Table of Contents
mzDB-processing
Purpose
Extracting peptidic signals (called “features”) from a file converted into the mzDB format.
Feature extraction algorithms
The FeatureExtractor algorithm is composed of four different extraction strategies:
- UnsupervisedFeatureExtractor (NYI)
- MS2DrivenFeatureExtractor
- PredictedTimeFeatureExtractor
- PredictedMzFeatureExtractor (NYI)
The selection of the strategy depends on the PutativeFeatures parameters.
Details on these different implementations are given in the following sections.
MS2 driven algorithm
This is the main peptide signals extraction algorithm. Every MS/MS event triggered by the spectrometer corresponds to one or more peptidic signal. Each event provides a set of information about the targeted precursor ion: the m/z ratio (assuming it is monoisotopic), the moment when the MS/MS has been triggered (usually not the maximum of the elution peak) and the charge state of the ion. The first and second information can be considered as close coordinates for the peptide signal on the LC-MS map. The charge state (z) can provide additional information to simplify the extraction of different isotopes of the features which are approximately separated by 1/z. For each MS/MS event:
- The runSlice containing the precursor m/z of the MS/MS event is retrieved (default window is 5 Da, more details in the mzDB documentation), as well as the following runSlice, in order to load into memory everything about the peptidic signal including the isotopes. The XIC for the MS/MS precursor mass can be then easily accessed with a user defined mass precision (default is 5ppm).
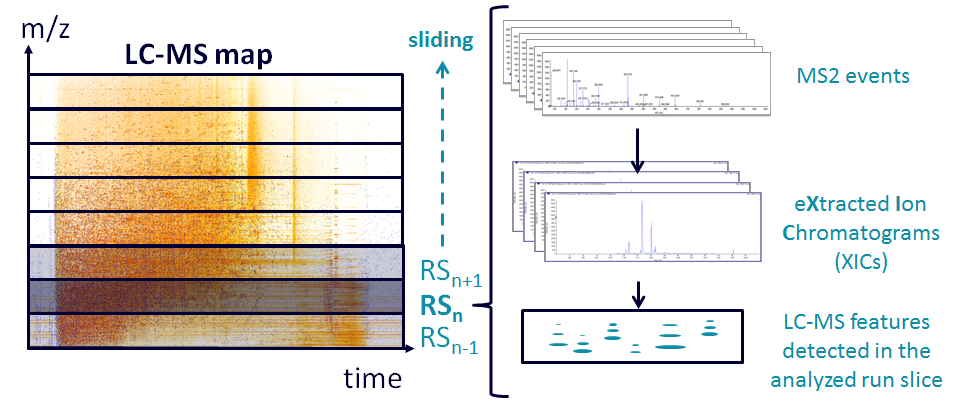
- The apex of the elution peak of the monoisotopic mass of the peptide does not exactly fit to the moment the MS/MS was triggered. Knowing that, the signal on the XIC is integrated on both sides of the moment the MS/MS was triggered (default value is 10 scans) to determine the ascendant slope and in order to find the apex. The integration of the signal is done by summing the intensities of n isotopes, n being a user-defined value (default value is 3, including the monoisotopic peak).

- For each isotopic profile, the intensities are extracted allowing gaps (default value is 1) until a minimal intensity is reached. This minimal intensity is defined as a ratio of the detected apex intensity (default value is 0.001%). Only one extraction is done per spectrum, hence reducing the extraction time (theoretically).

- The peak is detected on the extracted signal corresponding to the isotope signal with the highest relative intensity predicted by the averagine (most of the time it corresponds to the monoisotopic peak, in conventional conditions such as trypsic digestion). The limits of this peak are used to tune all the limits of the isotopes (elution peaks). To do so, two different algorithms are being tested:
- “Basic” algorithm: applying a Savitsky-Golay smoothing then looking for the local highest point.
- Wavelet-based algorithm: using multiple wavelet transformed curves to determine the position of the peaks
- The last step consists in extracting the peptide signals containing a strong overlap with the previously extracted signal (especially with the first two isotopes).



The extraction of all the signals corresponding to MS/MS events is made in a single iteration on all the runSlices of a mzDB file. Also all the peptide signals which mass are contained in the runSlice are detected simultaneously.
PredictedTimeExtractor algorithm
This algorithm is used for cross-assignment, when a peptidic signal is detected in a file but does not have an equivalent signal in another (frequently in DDA). In this case, the algorithm will try to extract some signal from the file where the signal has not been found. The aim of this algorithm is to reduce the number of missing values.
- Extracting a 4-minutes XIC (user-defined value) around:
- the time predicted by the alignment
- the ratio m/z of the isotope with the highest intensity predicted by the averagine (which is estimated from the mean value of the m/z of the observed signals in other conditions)
- Peaks are detected with the wavelet based algorithm (usually better for a signal made of hundreds of peaks) and limits of time are determined. The isotopic profiles are extracted for each spectrum using the method as in the MS2Driven algorithm. Many peptide signals can be detected and need to be filtered in order to find the best match with the signals in other conditions.
- To do so, we verify beforehand that :
- The chromatographic elution peaks of the monisotopic mass are really corresponding to monoisotopic masses: i.e., if no elution peak P is present before the considered monoisotopic mass M that has a difference of mass equal to 1.0027/z (z being the charge of M), having a distance apex-to-apex (P vs. M) lower than a user-defined threshold of number of cycles (default value is 5), a Pearson correlation higher than a user-defined threshold (default value is 0.7) and finally a P/M area ratio agreeing with the predicted value for P using averagine.
- If needed a filter of the duration of a peptide signal (which is usually peptide-specific)
- Considering the signals close to each other in time (elution time at the apex vs. predicted time)
- Consider the signals close to each other in m/z ratio
Page Tools
