Table of Contents
Retention Time Alignment
Major steps followed by the Retention Time Alignment algorithm

- Collect species from the reference User context and predict their retention time using an external utility and store them in reference species properties
- For each identification gathered under the reference User context do:
- Collect species to align from the identification
- Constitute several pair groups between collected reference species & species to align. A pair group contains 2 groups of species having same sequence & calculated mass (group1 has species from reference User context and group2 has species to align from identifications).
- Compute one (or several) representative value(s) for group1 & group2 for each pair group
- Compute linear regression between representative values
- Store linear regression coefficients & reference context name in identification properties
In more details...

- Species Retention Time of the reference UserContext are predicted with NETPrediction v2.2.3378 utility using Kangas method (click here for more details). NETPrediction utility only uses species sequences to predict a Normalized Elution Time (NET) value.
- First, a list of 'reference' species is built
- The reference species list doesn't contain any species with PTMs
- If several species exist with redundant sequences, the best score species is retained
- Then, the corresponding sequences are exported in order to be used by the NETPrediction utility
- The predicted NET are converted to retention time using user-defined parameters (duration & delay).
- The user can decide to exclude predicted values too far from the others:
- The average absolute deviation (between RT & predicted RT) is computed and all predicted RT far from this average value about a given user-defined threshold are excluded.
- The predicted RT values are then stored as properties in the reference species.
- For each identification existing under the reference User context (not necessary directly under the reference user context):
- All the final child species to align are collected
- Several pair groups are created using reference species & species to align. A pair group contains 2 groups of species having same sequence & calculated mass (group1 has species from reference User context and group2 has species to align from identifications).
- For each group, according the statistical method choosen by the user, one or several representative RT value(s) is(are) calculated.
- Each 'Group1' always contains one species because species/protein grouping has been executed on the reference context and suppressed sequence/calculated mass redundancy.
- 'Group2' may contain one or more species. One or several representative value(s) is(are) calculated from one of following statistical methods:
- Average
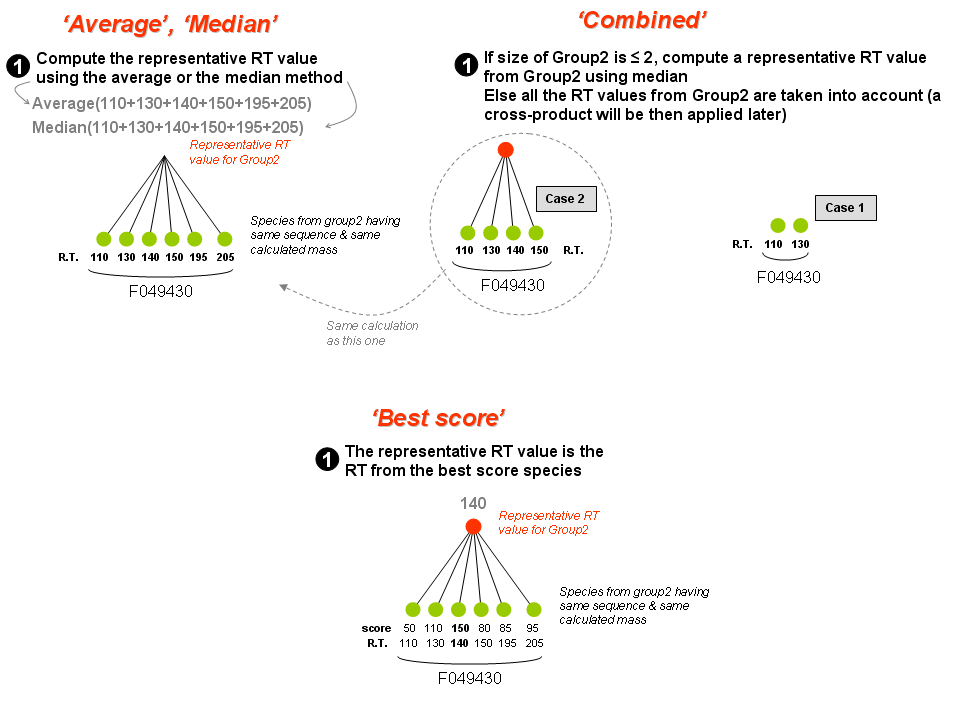
- Median
- Cross-product
- Combined
- Best score
- Compute linear regression between pair groups
- Store linear regression coefficients (slope/intercept) + reference context name in the identification properties
